Les inhibiteurs du récepteur V2 (V2R) de l’arginine-vasopressine (AVP) sont des composés thérapeutiques primordiaux pour traiter une hyponatrémie consécutive à une insuffisance cardiaque, une hypertension ou une cirrhose hépatique, mais aussi pour traiter certaines formes de polykystose rénale. Aujourd’hui deux molécules sont sur le marché, le Tolvaptan (TVP) et le Conivaptan. Alors que le TVP est sélectif du V2R, un traitement chronique conduit à des effets indésirables, en particulier une toxicité hépatique. Le Conivaptan, utilisé chez les patients hospitalisés pour une situation d’hyponatrémie liée à une insuffisance cardiaque, n’est pas sélectif du V2R car il possède une forte affinité pour le récepteur V1a de l’AVP.
Découvrir et développer de nouveaux composés avec une meilleure sélectivité et sans effet secondaire est donc crucial. Dans cette optique, le design rationnel de nouvelles drogues basé sur les structures expérimentales des récepteurs couplés aux protéines G est une approche performante. Cependant, jusqu’à présent, l’absence de structures du V2R lié à des inhibiteurs a empêché le développement de cette stratégie.
Après avoir publié et décrit les structures des conformations actives du V2R lié à l’hormone naturelle AVP et à deux partenaires canoniques de signalisation, la protéine Gs et la barrestin1, l’équipe IGF « Pharmacologie et biologie structurale des protéines membranaires » animée par Bernard Mouillac et Sébastien Granier, en collaboration avec Gunnar Schulte et Julien Bous du Karolinska Institute de Stockholm, Nicolas Gilles du Commissariat à l’Energie Atomique (CEA) de Paris, et Charline Mary du Centre de Biologie Structurale (CBS) de Montpellier, viennent de déterminer les structures du V2R en complexe avec deux inhibiteurs, le composé thérapeutique de référence TVP et la toxine Mambaquarétine (MQ1) issue du venin de serpent mamba vert par cryo-microscopie électronique. Les deux ligands interagissent avec la poche de liaison orthostérique du V2R mais avec des différences substantielles. La petite molécule TVP pénètre plus profondément que la MQ1, et bloque le V2R par un contact direct avec le tryptophane 284 (interrupteur on/off) situé dans le domaine transmembranaire 6. La toxine (peptide de la famille dite de Kunitz) établit de multiples contacts avec les résidus du V2R extracellulaires et transmembranaires et inhibe le récepteur d’une façon très spécifique. En accord avec les propriétés pharmacologiques du TVP et de la MQ1, les deux structures représentent des conformations inactives du V2R. La comparaison de ces structures inactives avec les structures actives du V2R lié à l’AVP révèle les mécanismes moléculaires qui modulent l’activité du récepteur.
Ces résultats constituent les premières structures du V2R en complexe avec des inhibiteurs jamais décrites. Leur comparaison avec les structures actives (en présence d’activateurs et de partenaires de signalisation) va pouvoir guider le développement rationnel de nouvelles molécules pour améliorer les thérapies actuelles. Cette perspective est cruciale lorsqu’il s’agit de pathologies qui sont difficiles à gérer et qui sont accablantes pour les patients. De plus, la structure qui met en jeu le V2R et la mini-protéine MQ1 ouvre de nouvelles perspectives pharmacologiques pour traiter les maladies rénales et de l’équilibre hydrique.
Ce travail vient d’être publié dans la revue Nature Communications.
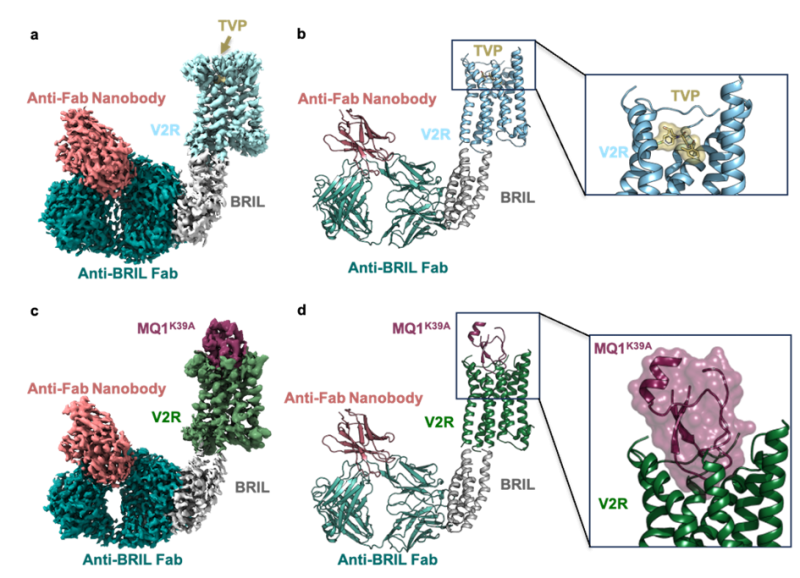
Structures inactives du V2R en complexe avec le tolvaptan and la mambaquarétine 1 (MQ1).
Dans les panneaux a et c, les cartes de densité de cryo-microscopie électronique sont montrées. Dans les panneaux b et d, les modèles 3D correspondants sont illustrés. Un zoom des complexes V2R-TVP et V2R-MQ1 (mutant K39A) est présenté sur la droite. Le module BRIL, le fragment d’anticorps (Fab) anti-BRIL et le “nanobody” anti-Fab sont utilisés comme partenaires d’échafaudage.
