Attribuer au moins une fonction à chaque gène et identifier les gènes qui concourent à une même fonction sont deux des buts fondamentaux de la génomique fonctionnelle. La co-expression fréquente d’un ensemble de gènes est l’indice que ces gènes sont fonctionnellement liés, c’est-à-dire qu’ils participent à au moins un même processus biologique. En accord avec la définition classique d’un module fonctionnel par Hartwell et al. (From molecular to modular cell biology. Nature, 1999, 402, C47–52), les méthodes permettant d’identifier de tels modules fonctionnels dans des réseaux de co-expression génique font toutes l’hypothèse que les membres d’un module ont plus de relations entre eux qu’avec les membres d’autres modules. Cette hypothèse est vérifiable dans des réseaux comprenant peu de gènes et l’identification de modules fonctionnels est relativement aisée. Dès que la taille des réseaux étudiés augmente, cette hypothèse ne peut pas être facilement testée. Nous avons donc choisi de ne pas faire d’hypothèse sur la topologie des modules fonctionnels mais plutôt d’apprendre leur topologie à partir de modules fonctionnels connus. Cette connaissance peut ensuite être utilisée pour identifier de nouveaux modules fonctionnels, trouver de nouveaux membres de modules déjà identifiés ou identifier les gènes fonctionnellement liés à un gène donné.
Les modules fonctionnels (MF) connus que nous avons utilisés sont les 978 ‘’Processus Biologiques’’ (PB) comprenant entre 20 et 500 gènes, tels que définis dans la base de données Gene Ontology (GO). Nous avons également constitué un nombre équivalent de modules aléatoires (MA) de même taille par tirage au hasard dans l’ensemble des gènes du réseau. Nous avons ensuite calculé les valeurs de 12 paramètres caractérisant la topologie des modules étudiés : degré, connectivité du voisinage, plus court chemin, coefficient de clustering, centralité de proximité… Nous avons utilisé la technique LASSO (Least Absolute Shrinkage and Selection Operator) pour identifier six paramètres qui différenciaient au mieux MA et MF et avons réalisé une analyse discriminante linéaire pour trouver une combinaison linéaire de ces paramètres. Cette procédure a permis d’attribuer un score topologique (ScoreTopo) à chaque module. Ce score est élevé si la topologie du module ressemble à celle d’un PB connu et faible si elle ressemble à celle d’un MA. Parallèlement, nous avons utilisé la méthode de Wang (Wang JZ et al. (2007) A new method to measure the semantic similarity of GO terms. Bioinformatics, 23, 1274–81.) pour définir un score fonctionnel (ScoreFun) pour chaque module. Ce score est basé sur la distance des annotations des gènes du module dans l’arbre reliant les termes GO. Ce score est élevé si les gènes qui constituent le module sont fonctionnellement proches et faible dans le cas contraire. La combinaison des 2 scores permet de mesurer à quel point un module candidat est constitué de gènes aussi fonctionnellement apparentés que ceux des PB GO.
Le nombre de modules candidats dans un réseau d’environ 20.000 gènes est beaucoup trop grand pour envisager une exploration exhaustive ; nous avons choisi d’utiliser un algorithme génétique que nous avons nommé TopoFun. ScoreTopo et ScoreFun ont été combinés dans une fonction d’évaluation (fitness function) capable de classer différents modules. En partant d’une population de modules candidats, en les faisant évoluer par mutation (mutation ponctuelle, insertion, délétion, recombinaison) et en les sélectionnant grâce à la fonction d’évaluation, l’algorithme converge après quelques milliers d’itérations vers un module qui représente une excellente solution (à défaut d’avoir la certitude qu’il s’agit de la meilleure).
Afin de confirmer l’intérêt de notre méthode, nous sommes partis de PB connus et avons essayé de les ‘’améliorer’’ en cherchant dans le réseau de co-expression de nouveaux gènes, non annotés par ce PB, mais dont la co-expression avec les membres connus de ce PB suggère qu’ils concourent à la même fonction. En utilisant une base de données indépendante (STRING), nous avons vérifié que les nouveaux gènes ainsi obtenus étaient significativement liés fonctionnellement à ceux déjà connus. Enfin, nous avons évalué les modules trouvés par WGCNA (Weighted Gene Co-expression Network Analysis), la méthode de référence en analyse de réseaux de co-expression. Ces modules ont une excellente topologie, ‘’supérieure’’ à celles des PB connus. Ils souffrent cependant d’une faible cohérence fonctionnelle et ressemblent plus à des MA de ce point de vue. Nous avons donc utilisé TopoFun pour les améliorer et avons produit des modules présentant une topologie très légèrement améliorée mais surtout une cohérence fonctionnelle bien supérieure.
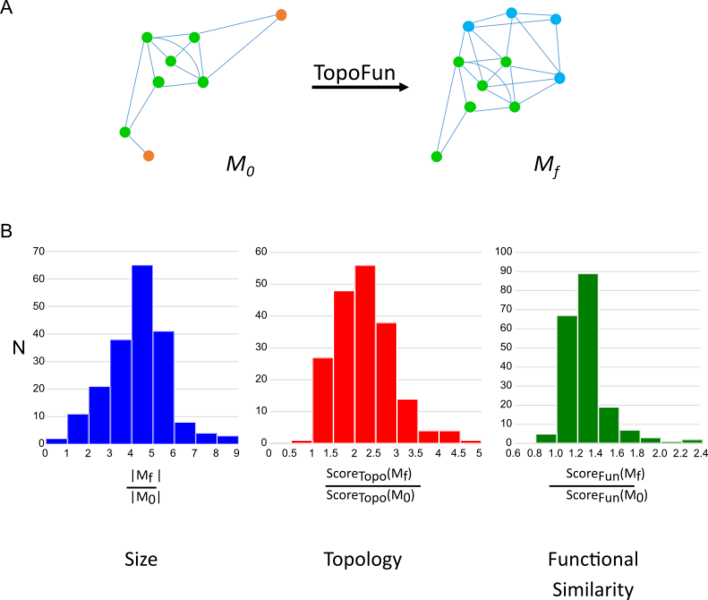
En partant d’un ensemble de gènes donnés (A, M0), TopoFun élimine les gènes les moins co-exprimés (orange) et identifie de nouveaux gènes (bleu) co-exprimés avec ceux du module original, tout en améliorant (B) la taille, la topologie et la similarité sémantique du module final (Mf).
TopoFun: a machine learning method to improve the functional similarity of gene co-expression modules. Janbain A, Reynès C, Assaghir Z, Zeineddine H, Sabatier R, Journot L. NAR Genom Bioinform, 2021.
