The functional annotation of each gene and the identification of genes involved in each function are two basic goals of functional genomics. The frequent co-expression of members of a gene set is an indication that the constituting genes are functionally related and participate to one or more common biological process(es). Following the classical definition of functional modules by Hartwell and colleagues (From molecular to modular cell biology. Nature, 1999, 402, C47–52), the methods aimed at identifying functional modules all assume that the members of a module have more links with one another than they have with genes in other modules. This hypothesis can be verified in networks comprising a limited number of genes and the identification of functional modules is then relatively easy. When the number of genes increases, this hypothesis is more difficult to test. We decided to make no hypothesis about the functional modules topology; we rather learned their topology from known functional modules using machine learning. This knowledge is then useful to identify new functional modules, to find new members of known functional modules, and to identify functionally related genes for a given seed gene.
We studied the 978 Gene Ontology Biological Processes (GO-BPs) with 20-500 genes as prototypic functional modules (FMs). We generated an equivalent number of random modules (RMs) made of equivalent numbers of randomly sampled genes. We computed 12 topological descriptors of the modules topology: degree, neighborhood connectivity, shortest path, clustering coefficient, closeness centrality… Using LASSO (Least Absolute Shrinkage and Selection Operator), we selected the six topological descriptors that best discriminated FMs and RMs. Using the selected topological descriptors, we performed LDA (Linear Discriminant Analysis) to construct a topological score (ScoreTopo) that predicted the type of a module, random-like or functional-like. Based on the work of Wang and colleagues (A new method to measure the semantic similarity of GO terms. Bioinformatics, 2007, 23, 1274–81), we designed a functional similarity score (ScoreFun) based on the distance in the GO tree of the annotations of the genes that constitute a module; the score is high if the module’s genes are functionally close and low otherwise. ScoreTopo and ScoreFun estimate to which extent a module is made of genes as functionally related as those of GO-BPs.
The number of candidate functional modules in a network of about 20,000 genes is much too high for a systematic assessment. We combined ScoreTopo and ScoreFun in a fitness function that ranked candidate modules and we designed a genetic algorithm we named TopoFun to explore the co-expression network. Starting from a population of candidate modules to which we applied mutations (point mutations, insertions, deletions, crossing-over) and selection using the fitness function, TopoFun converged after a few thousands of iterations towards a module representing a very good solution (although we cannot prove it is the best one).
To illustrate the use of TopoFun, we started from a subset of the Gene Ontology Biological Processes (GO-BPs) and showed that TopoFun efficiently retrieved genes that we omitted, and aggregated a number of novel genes to the initial GO BP while improving module topology and functional similarity. Using an independent protein-protein interaction database (STRING), we confirmed that the novel genes gathered by TopoFun were functionally related to the original gene set. Finally, we evaluated the functional modules produced by WGCNA (Weighted Gene Co-expression Network Analysis), the reference method in network algorithms. WGCNA modules display an exceptional topology, better than that of known GO-BPs. However, they lack functional coherence and are not significantly different from RMs from that point of view. We used TopoFun to improve the WGCNA modules functional similarity and produced modules with a slightly improved topology and much higher functional consistency.
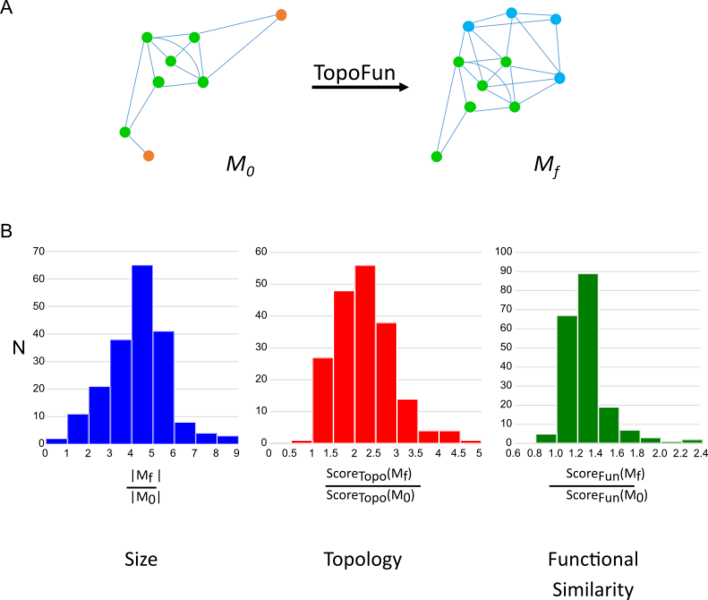
Starting from a set of given genes (A, M0), TopoFun eliminates the least co-expressed genes (orange) and identifies new genes (blue) co-expressed with those in the original module, while improving (B) the size, topology and semantic similarity of the final module (Mf).
TopoFun: a machine learning method to improve the functional similarity of gene co-expression modules. Janbain A, Reynès C, Assaghir Z, Zeineddine H, Sabatier R, Journot L. NAR Genom Bioinform, 2021.
